Hereditary Angioedema (HAE), atau angioedema herediter, adalah penyakit genetik langka yang menyebabkan pembengkakan berulang pada jaringan tubuh di bawah kulit maupun di bawah lapisan dalam organ. Tidak seperti reaksi alergi biasa, pembengkakan pada HAE terjadi tanpa disertai gatal-gatal (urtikaria), dan seringkali membingungkan pasien maupun dokter yang tidak familiar dengan kondisi ini.1-2
Hereditary Angioedema merupakan penyakit autosomal dominan yang disebabkan oleh kekurangan atau disfungsi protein C1-inhibitor. Angka kejadiannya diperkirakan sekitar 1 dari 50.000 orang di seluruh dunia — tergolong penyakit langka, namun dampaknya bisa sangat berat bahkan mengancam jiwa. HAE sering salah didiagnosis selama bertahun-tahun. Rata-rata waktu antara munculnya gejala pertama hingga diagnosis HAE tegak adalah sekitar 13 tahun.1
Penyakit HAE ini dikenal sejak hampir 150 tahun lalu, namun variannya terus berkembang. Saat ini HAE dibagi menjadi tiga tipe utama: HAE Tipe 1 (defisiensi C1-INH), HAE Tipe 2 (C1-INH tidak berfungsi normal), dan HAE dengan C1-INH normal. Akar masalah HAE terletak pada kelainan gen. Pada HAE Tipe 1 dan 2, terjadi mutasi pada gen SERPING1 yang bertugas mengkodekan protein C1-inhibitor (C1-INH). C1-INH adalah “penjaga keseimbangan” dalam sistem komplemen dan sistem kontak darah. Ketika C1-INH tidak mencukupi atau tidak berfungsi baik, terjadi produksi berlebih dari sebuah zat yang disebut bradikinin — hormon yang menyebabkan pembuluh darah bocor sehingga cairan merembes ke jaringan sekitarnya dan terjadilah pembengkakan. Lebih dari 150 jenis mutasi berbeda telah diidentifikasi pada gen SERPING1. Pada Tipe 1, mutasi menyebabkan protein C1-INH tidak dapat disekresikan dengan baik sehingga kadarnya rendah. Pada Tipe 2, protein C1-INH terbentuk dalam jumlah normal atau bahkan berlebih, namun tidak berfungsi sebagaimana mestinya.1
Hereditary angioedema diwariskan secara autosomal dominan — cukup satu salinan gen bermutasi untuk menyebabkan penyakit. Sekitar 70% kasus memiliki riwayat keluarga dengan HAE Sekitar 25% kasus merupakan mutasi baru (de novo) tanpa riwayat keluarga. Gejala biasanya mulai muncul pada dekade pertama atau kedua kehidupan. Faktor pencetus serangan sangat beragam. Stres emosional, trauma fisik, infeksi, perubahan hormon, prosedur gigi atau operasi, serta konsumsi obat-obatan tertentu seperti ACE inhibitor dan estrogen (misalnya dari kontrasepsi oral) dapat memicu episode akut. Pada sekitar 40% serangan, pemicunya tidak dapat diidentifikasi secara jelas.3
Pembengkakan pada HAE dapat muncul di berbagai lokasi tubuh dan gejalanya sangat bergantung pada lokasi tersebut. Tiga area utama yang terkena adalah kulit, saluran cerna, dan saluran napas.1
- Kulit
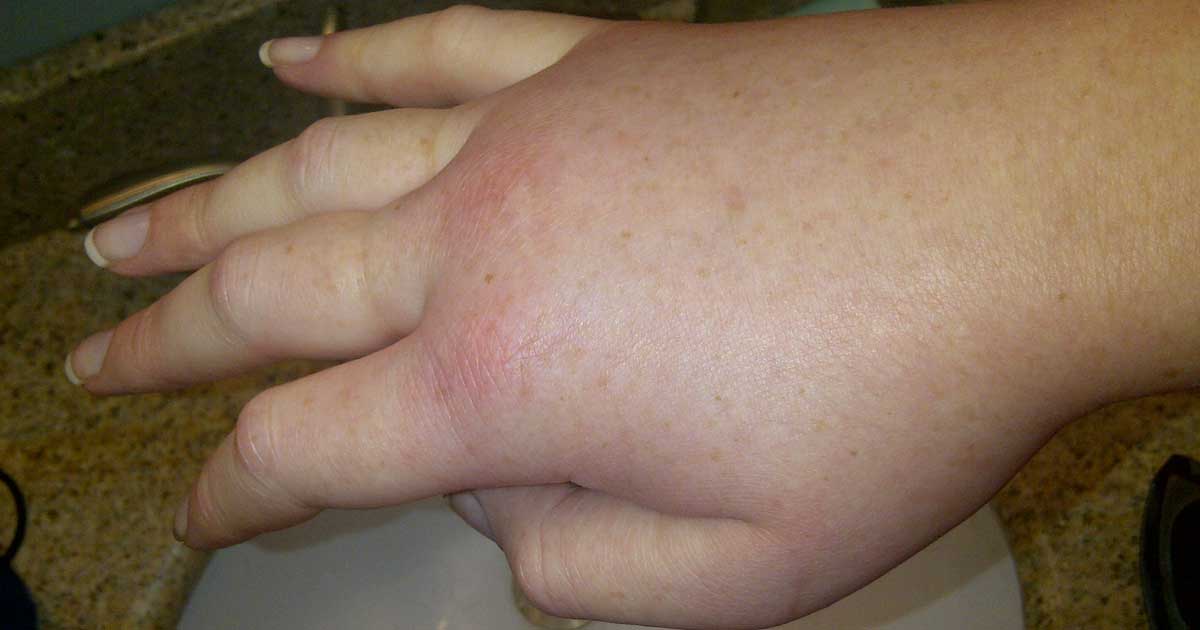
Pembengkakan subkutan (di bawah kulit) paling sering terjadi, mengenai tangan, kaki, wajah, alat kelamin, dan bibir. Bengkak ini tidak disertai rasa gatal, namun bisa sangat nyeri dan mengganggu penampilan. Lebih dari 90% pasien mengalami gejala kulit ini.1
- Saluran Cerna
Edema pada dinding usus memicu nyeri perut hebat, mual, muntah, bahkan diare. Gejala ini seringkali disalahartikan sebagai apendisitis atau penyakit perut lainnya, sehingga banyak pasien HAE yang pernah menjalani operasi perut yang tidak perlu. Sekitar 84% pasien mengalami gejala gastrointestinal.1
- Saluran Napas (Laring)
Pembengkakan pada laring (tenggorokan bagian dalam) dapat menyumbat jalan napas dan menyebabkan kematian jika tidak segera ditangani. Untungnya, keterlibatan laring hanya terjadi pada sekitar 1–3% dari seluruh serangan, namun risikonya tetap harus selalu diwaspadai. Pembengkakan pada lidah, tenggorokan, atau suara yang tiba-tiba serak dapat menandakan edema laring. Ini adalah kondisi darurat yang memerlukan penanganan segera di unit gawat darurat.1-2
Serangan umumnya berlangsung selama 2 hingga 5 hari, perlahan memburuk kemudian mereda sendiri bahkan tanpa pengobatan. Sebelum serangan, banyak pasien mengalami gejala prodromal berupa ruam tidak gatal berbentuk cincin yang disebut erythema marginatum.3
Diagnosis HAE memerlukan kombinasi antara riwayat klinis, riwayat keluarga, dan pemeriksaan laboratorium. Tidak ada pemeriksaan tunggal yang bisa langsung menyimpulkan HAE — semuanya perlu diintegrasikan. Pemeriksaan laboratorium dimulai dari kadar C4, yang sering digunakan sebagai tes skrining awal. C4 hampir selalu rendah pada pasien HAE Tipe 1 dan 2, bahkan ketika tidak sedang terjadi serangan aktif. Jika C4 rendah, dilanjutkan dengan pemeriksaan kadar dan fungsi C1-inhibitor. Pada HAE Tipe 1, keduanya rendah; pada Tipe 2, kadar C1-INH normal atau tinggi tetapi fungsinya menurun. Pemeriksaan C1q kadang ditambahkan untuk menyingkirkan angioedema yang didapat (acquired angioedema), terutama pada pasien yang baru bergejala di usia dewasa, karena pada angioedema yang didapat, C1q biasanya rendah, sementara pada HAE nilainya normal.3
Penanganan HAE berbeda total dari angioedema akibat alergi. Antihistamin, kortikosteroid, dan epinefrin — yang andal untuk reaksi alergi — tidak efektif untuk HAE karena mekanismenya berbeda: HAE dimediasi oleh bradikinin, bukan histamin. Hal ini juga yang menyebabkan banyak pasien tidak membaik saat mendapat penanganan standar di UGD. Pengobatan HAE terbagi menjadi dua strategi utama: terapi akut (saat serangan terjadi) dan profilaksis jangka panjang (mencegah serangan berulang).3-4
Untuk terapi akut, beberapa pilihan yang terbukti efektif antara lain konsentrat C1-inhibitor (diberikan secara intravena atau subkutan), icatibant (antagonis reseptor bradikinin B2), dan ecallantide (inhibitor plasma kallikrein). Obat-obatan ini bekerja langsung memutus jalur yang menyebabkan kebocoran pembuluh darah. Untuk profilaksis jangka panjang — terutama pada pasien dengan serangan yang sering dan berat — tersedia pilihan seperti konsentrat C1-INH subkutan, lanadelumab (antibodi monoklonal yang menghambat plasma kallikrein), serta berotralstat (inhibitor kallikrein oral). Terapi-terapi baru ini telah merevolusi penanganan HAE dan secara signifikan mengurangi frekuensi serangan. Perkembangan terapeutik terbaru memungkinkan banyak pasien HAE hidup nyaris normal — dengan frekuensi serangan yang sangat berkurang atau bahkan nol.4
Kesimpulan
Hereditary angioedema adalah penyakit langka yang nyata dan serius, namun sering tersembunyi di balik diagnosis yang salah selama bertahun-tahun. Jika Anda atau anggota keluarga mengalami pembengkakan berulang yang tidak gatal, tidak merespons antihistamin, dan terutama jika ada anggota keluarga lain dengan keluhan serupa, segera konsultasikan ke dokter spesialis alergi-imunologi atau penyakit dalam.1-4
Penulis :

Referensi
- Sinnathamby ES, Issa PP, Roberts L, Norwood H, Malone K, Vemulapalli H, et al. Hereditary angioedema: diagnosis, clinical implications, and pathophysiology. Adv Ther. 2023;40(3):814-827. doi:10.1007/s12325-022-02401-0.
- Uminski K, Betschel S, Goodyear D. Hereditary angioedema. CMAJ. 2025;197(15):E417-E418. doi:10.1503/cmaj.241815.
- Abdulkarim A, Craig TJ. Hereditary angioedema. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan [updated 2023 May 1].
- Uminski K, Goodyear D, Betschel S. Therapeutic advances in hereditary angioedema: a focus on present and future options. Adv Ther. 2025;42(12):5879-5895. doi:10.1007/s12325-025-03382-6.